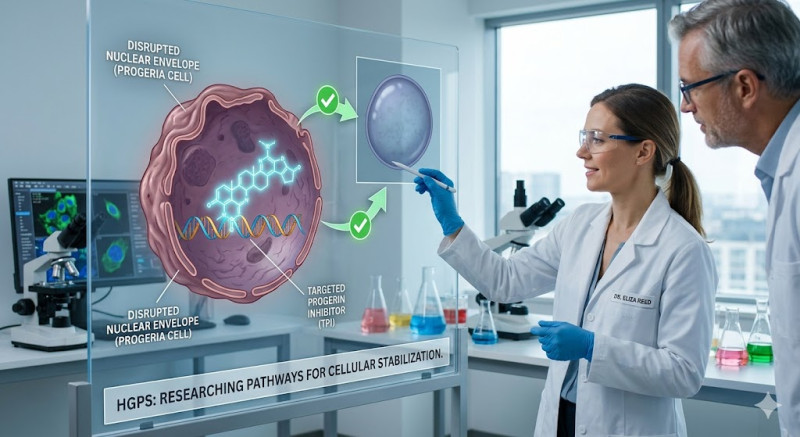
Hutchinson-Gilford Progeria Syndrom (HGPS), vanligvis bare kalt progeria, er en ekstremt sjelden og dødelig genetisk tilstand preget av en akselerert og dramatisk aldringsprosess hos barn. Navnet stammer fra gresk og betyr "før alderdom". Tilstanden rammer omtrent én av fire til åtte millioner nyfødte over hele verden. Sykdommen skyldes ikke en arvelig defekt som overføres fra foreldrene, men oppstår som en spontan de novo-mutasjon (en nyoppstått mutasjon) i genet LMNA. Dette genet koder for proteinet Lamin A, som fungerer som et strukturelt stillas for cellekjernen.
Sykdomstegn og klinisk forløp
Barn med progeria ser helt normale ut ved fødselen, men i løpet av det første leveåret begynner de første tegnene å vise seg. Veksten flater ut, og de utvikler et karakteristisk utseende: et relativt stort hode i forhold til ansiktet, tynn og gjennomsiktig hud med synlige årer, hårtap (alopesi) som fører til total skallethet, samt tap av underhudsfett. De utvikler også typiske alderdomssykdommer i svært ung alder, som stive ledd, hoftedislokasjoner og alvorlig, progressiv åreforkalkning (aterosklerose). Til tross for at kroppen eldes i et voldsomt tempo, påvirkes ikke barnets mentale utvikling; de forblir kognitivt og emosjonelt som andre barn på sin egen alder.
Behandling og levealder
Gjennomsnittlig levealder for et barn med progeria er rundt 14,5 år, og dødsårsaken er nesten alltid hjerteinfarkt eller hjerneslag som følge av den aggressive åreforkalkningen. Nevrobiologisk og medisinsk forskning har gjort store fremskritt de siste årene. Oppdagelsen av LMNA-mutasjonen i 2003 banet vei for målrettet behandling. Den unormale mutasjonen produserer et defekt protein kalt progerin, som samler seg opp i cellekjernen og ødelegger cellens stabilitet. I dag finnes det medisiner, som farnesyltransferase-inhibitorer (f.eks. lonafarnib), som kan bidra til å redusere opphopningen av progerin. Dette kan forlenge levetiden og forbedre livskvaliteten, selv om en endelig kur ennå ikke eksisterer.